
 2023-09-07
2023-09-07
作者:Orlando Lopez
美国食品药品监督管理局(FDA)于2023年8月1日在其网站上发布了一份新的警告信。这份日期为2023年7月28日的文件内容是关于2022年11月22日-12月2日在萨南德(印度古吉拉特邦)的一家药品生产工厂进行的一次审查。
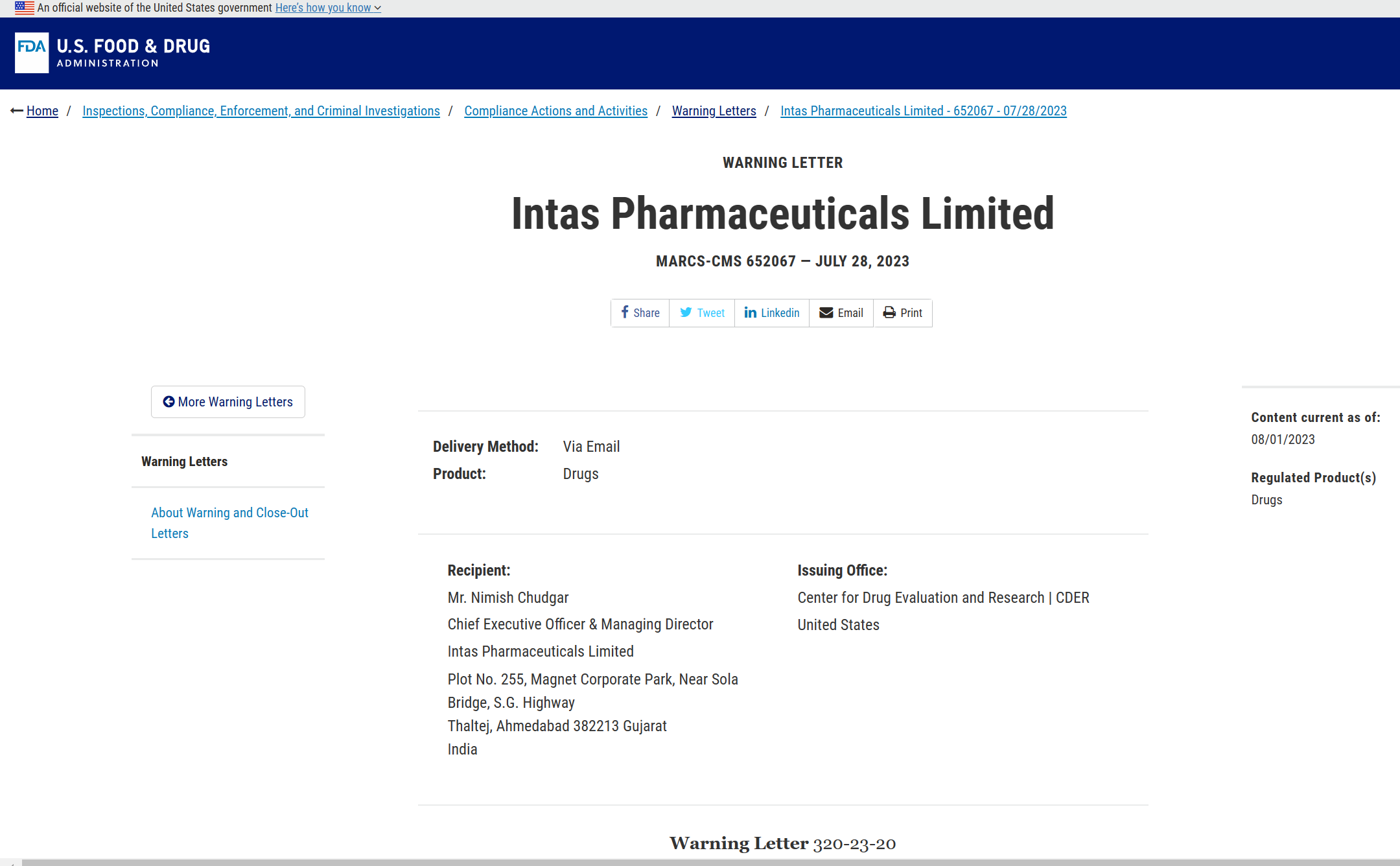
图片来源:FDA官网
该警告信提及数据、质量以及实验室关键控制程序缺失等多个问题。
警告信特别指出了在质控监督方面,与计算机系统相关的违规行为:“贵公司质量控制部门未能履行其职责,以确保药品符合CGMP,并确保符合既定的性质、强度、质量和纯度标准(21 CFR 211.22)。你们应采取措施确保关于你们工厂生产药品质量的数据的可靠性。我们检查发现了严重的偏差,包括不限于:原始CGMP文件监管不充分、计算机化系统控制不足、实验室调查不充分和色谱序列中断。”
2022年12月的美国FDA 483表格中的第10项观察结果,强调了上述与计算机系统相关的违规行为。

图片来源:FDA官网
观察结果10
没有针对计算机或相关系统采取适当的控制措施,以确保只有授权人员才能对主生产控制记录或其他记录进行更改。
作为PQ160193的一部分,已对183台生产设备和系统进行了数据完整性控制评估。评估报告于2018年3月5日获得批准,发现149台设备需要升级用户访问控制和权限,76台设备需要限制用户更改时间,113台设备需要升级电子数据保存功能,119台设备需要升级审计跟踪功能。根据评估报告,需要在质量体系中有明确的记录,以确保通过软件升级或实施过渡控制措施来进行适当控制。
目前,尚未对实验室设备进行类似的数据完整性控制评估。滴定仪器是用于xx和测定测试的独立系统。该仪器可以储存电子结果数据,但该功能暂未使用。相反,该流程依赖纸质打印输出,但没有进行二次核查,所以无法确保所有打印输出都得到保留和报告。2022年11月22日,在废弃区发现了原始xx打印输出的废弃物。
电子批记录未配置以确保所有相关数据得到同步记录。

图片来源网络
除了向FDA提供常规的数据完整性相关信息和整改措施外,还需要提供以下信息:
一个综合性的评估和整改计划,可以确保质量保证部门具备有效履职的权力和资源。该评估还应包含但不限于以下内容:
FDA得出结论是:“质量体系无法充分确保数据的准确性和完整性,以支持贵司所生产药品的安全性、有效性和质量。” FDA建议根据GMP违规的性质,聘请一名专业的顾问。
我们必须理解,要建立和维持GMP合规系统,验证专家必须了解监管当局对系统的生命周期、维护和运行方面的期望。这些期望在软件开发标准以及法规要求和指南中都有所阐述。














